Erika McCarthy1, Şölen Ekesan1, and Darrin M. York1
1Laboratory for Biomolecular Simulation Research, Institute for Quantitative Biomedicine and Department of Chemistry and Chemical Biology, Rutgers University, Piscataway, NJ 08854, USA
In this tutorial, you will solvate the MTR1 ribozyme in explicit water starting from the crystal structure. The associated files can be downloaded from Files.tar.gz. The tutorial is structured as follows:
The necessary inputs and subsequent outputs for parts 1.1 and 1.2 are available in:
Files/input/build and Files/output/build
The inputs should be copied to your working directory. If you unable to produce an output for a given step, copy it from the output directory in order to proceed.
This tutorial contains slurm scripts suitable for the Expanse supercomputer, so environment specific modifications will need to be made by the user.
[user@cluster] mkdir MTR1_build
[user@cluster] cd MTR1_build
[user@cluster] cp -r Path2/Files/input/build/* ./ The crystal structure of MTR1 can be obtained from the Protein Data Bank under the ID 7v9e.
You have been provided a set of representations to visualize various aspects of the crystal structure in MTR_crystalreps.txt:
# MTR1 crystal representations
display depthcue off
mol showrep top 0 on
mol selection {all}
mol representation Lines
mol addrep top
# Ions and water (vmd thinks the ligand is solvent, so exclude it)
mol selection {solvent and (not resname GUN)}
mol representation CPK
mol addrep top
# Active site residues
mol selection {resid 10 45 63 101}
mol representation licorice
mol addrep top[user@local] vmd 7v9e.pdbYou will see various crystallographic waters and ions that we will ultimately clean from the pdb file before building the system. You will also see that the ribozyme contains two loops and a stem region that meet at a 3 way junction. Junctions are important in RNA structures because they involve bringing the negatively charged phosphates from the backbone close together. This is typically mediated by essential interactions with solvent. Notice that the active site, shown in licorice, is located at the junction.
This crystal captures the product state of the ribozyme. We see that adenine 63 is methylated at the N1 position. The ribozyme binds O6 methylated guanine to initiate the reaction, so the reactant state would have the methyl group on guanine and a standard adenine for 63. While in the product state N1 of guanine is protonated, the reactant state O6 methylated guanine does not have a proton at N1. We will hypothesize the source of that proton to be N3 position of C10, meaning the reaction starts with C10 protonated with a +1 charge.
PDB files contain many lines giving information about the structure, as well coordinate information for each resolved atom.More information regarding pdb format can be found here. The flags corresponding to atoms and their coordinates, which are the most relevant information for MD simulations, are "ATOM" and "HETATM" where ATOM corresponds main coordinate of the atom, HETATM correspond to coordinates of solvent and ligand atoms not part of the main structure.
This pdb file has anisotropic temperature factors reported in ANISOU lines. Since we will have to manually edit some lines in our pdb, we want to work with the minimalist version of the file. We will use the pdb4amber program to achieve that.
[user@cluster] pdb4amber --in 7v9e.pdb --out 7v9e_4amber.pdb
==================================================
Summary of pdb4amber for: 7v9e.pdb
===================================================
----------Chains
The following (original) chains have been found:
A
---------- Alternate Locations (Original Residues!))
The following residues had alternate locations:
None
-----------Non-standard-resnames
GUN
---------- Gaps (Renumbered Residues!)
gap of 8.021418 A between G 61 and G 63
---------- Missing heavy atom(s)
None[user@local] vmd -m 7v9e.pdb 7v9e_4amber.pdb Notice that the two structures are indistinguishable. pdb4amber removed introductory lines, ANISOU lines and CONECT lines at the end, and renumbered atoms and residues to be sequential (i.e. residue and/or atom numbers might be different between the two files). Residue 62 (previously 63) is the 1-methyl adenosine, which was not recognized as a standard RNA residue, causing a perceived gap between G 61 and G 63. This is not a problem. Next we will make some manual modifications to our pdb, so let's first make a new copy to work with.
[user@cluster] cp 7v9e_4amber.pdb 7v9e_prot.pdb The remaining REMARK lines would be important for other types of MD simulations such as crystal simulations, but won't be relevant in this exercise. So if desired they can be removed, but also keeping them will have no effect. The following text blocks have line numbers turned on in vim (:set nu) to help you navigate the file.
2 REMARK 290 SMTRY1 1 1.000000 0.000000 0.000000 0.00000
3 REMARK 290 SMTRY2 1 0.000000 1.000000 0.000000 0.00000
4 REMARK 290 SMTRY3 1 0.000000 0.000000 1.000000 0.00000
5 REMARK 290 SMTRY1 2 -1.000000 0.000000 0.000000 0.00000
6 REMARK 290 SMTRY2 2 0.000000 -1.000000 0.000000 0.00000
7 REMARK 290 SMTRY3 2 0.000000 0.000000 1.000000 49.96950
8 REMARK 290 SMTRY1 3 -1.000000 0.000000 0.000000 0.00000
9 REMARK 290 SMTRY2 3 0.000000 1.000000 0.000000 0.00000
10 REMARK 290 SMTRY3 3 0.000000 0.000000 -1.000000 49.96950
11 REMARK 290 SMTRY1 4 1.000000 0.000000 0.000000 0.00000
12 REMARK 290 SMTRY2 4 0.000000 -1.000000 0.000000 0.00000
13 REMARK 290 SMTRY3 4 0.000000 0.000000 -1.000000 0.00000 Next we need to remove any phosphates at the 5' ends of nucleic acid chains. AMBER has distinct description for 5' and 3' residues where they are capped at O5' and XX atoms, respectively. The capping of the 5' end requires there to be no phosphate atoms.
2 ATOM 1 P C A 1 36.782 21.325 44.826 1.00 96.64 P
3 ATOM 2 OP1 C A 1 36.892 20.457 43.625 1.00 89.80 O
4 ATOM 3 OP2 C A 1 35.628 21.157 45.749 1.00 78.55 O Next we'll "protonate" C10. Since a protonated cytosine is not a standard residue we will use external parameters which we have named as CX.
ATTENTION: You need to maintain the pdb format where each block of information has a specific column range in the file, otherwise the file will cause errors when building the box in LEaP. Residue names correspond to columns 18 to 20,left aligned (i.e. start filling from 18):
260 ATOM 262 P CX A 9 18.771 0.638 43.621 1.00 55.52 P
261 ATOM 263 OP1 CX A 9 19.026 -0.826 43.656 1.00 74.66 O
262 ATOM 264 OP2 CX A 9 19.830 1.583 44.056 1.00 70.25 O
263 ATOM 265 O5' CX A 9 18.387 1.029 42.130 1.00 55.31 O
264 ATOM 266 C5' CX A 9 17.258 0.451 41.499 1.00 47.08 C
265 ATOM 267 C4' CX A 9 16.859 1.235 40.281 1.00 48.52 C
266 ATOM 268 O4' CX A 9 16.281 2.504 40.665 1.00 44.83 O
267 ATOM 269 C3' CX A 9 18.003 1.628 39.361 1.00 44.20 C
268 ATOM 270 O3' CX A 9 18.443 0.567 38.545 1.00 42.92 O
269 ATOM 271 C2' CX A 9 17.417 2.801 38.599 1.00 45.04 C
270 ATOM 272 O2' CX A 9 16.550 2.358 37.563 1.00 50.64 O
271 ATOM 273 C1' CX A 9 16.599 3.485 39.695 1.00 46.71 C
272 ATOM 274 N1 CX A 9 17.356 4.568 40.353 1.00 47.41 N
273 ATOM 275 C2 CX A 9 17.526 5.751 39.645 1.00 47.12 C
274 ATOM 276 O2 CX A 9 17.052 5.815 38.499 1.00 46.70 O
275 ATOM 277 N3 CX A 9 18.205 6.764 40.225 1.00 43.87 N
276 ATOM 278 C4 CX A 9 18.701 6.624 41.456 1.00 46.67 C
277 ATOM 279 N4 CX A 9 19.363 7.668 41.963 1.00 39.22 N
278 ATOM 280 C5 CX A 9 18.528 5.423 42.206 1.00 49.00 C
279 ATOM 281 C6 CX A 9 17.859 4.425 41.614 1.00 49.69 C
280 ATOM 282 H5' CX A 9 16.517 0.434 42.124 1.00 56.60 H
281 ATOM 283 H5'' CX A 9 17.470 -0.459 41.237 1.00 56.60 H
282 ATOM 284 H4' CX A 9 16.194 0.731 39.786 1.00 58.32 H
283 ATOM 285 H3' CX A 9 18.752 1.941 39.891 1.00 53.14 H
284 ATOM 286 H2' CX A 9 18.103 3.391 38.250 1.00 54.15 H
285 ATOM 287 HO2' CX A 9 16.855 2.616 36.823 1.00 60.87 H
286 ATOM 288 H1' CX A 9 15.781 3.844 39.317 1.00 56.16 H
287 ATOM 289 H41 CX A 9 19.699 7.621 42.753 1.00 47.16 H
288 ATOM 290 H42 CX A 9 19.452 8.387 41.500 1.00 47.16 H
289 ATOM 291 H5 CX A 9 18.866 5.336 43.069 1.00 58.90 H
290 ATOM 292 H6 CX A 9 17.735 3.623 42.068 1.00 59.73 H Next we will "mutate" the methylated adenine, 1MA, back to a standard adenine. This requires changing residue name from 1MA to A, and deleting the methyl atoms. We'll start with deleting the atoms so we have less atoms to rename residue names of.
1944 ATOM 1946 P A A 62 22.724 23.209 43.994 1.00 70.56 P
1945 ATOM 1947 OP1 A A 62 23.291 23.505 45.361 1.00 73.73 O
1946 ATOM 1948 OP2 A A 62 23.176 24.033 42.821 1.00 88.65 O
1947 ATOM 1949 O5' A A 62 22.969 21.684 43.646 1.00 63.06 O
1948 ATOM 1950 C5' A A 62 24.006 20.974 44.301 1.00 68.95 C
1949 ATOM 1951 C4' A A 62 23.489 20.121 45.431 1.00 58.78 C
1950 ATOM 1952 O4' A A 62 22.595 19.115 44.876 1.00 60.31 O
1951 ATOM 1953 C3' A A 62 24.569 19.343 46.178 1.00 56.81 C
1952 ATOM 1954 O3' A A 62 24.128 19.101 47.511 1.00 55.15 O
1953 ATOM 1955 C2' A A 62 24.610 18.029 45.411 1.00 51.57 C
1954 ATOM 1956 O2' A A 62 25.177 16.949 46.126 1.00 52.34 O
1955 ATOM 1957 C1' A A 62 23.117 17.826 45.125 1.00 54.19 C
1956 ATOM 1958 N9 A A 62 22.827 16.957 43.975 1.00 54.44 N
1957 ATOM 1959 C8 A A 62 23.376 17.668 42.954 1.00 52.77 C
1958 ATOM 1960 N7 A A 62 23.192 16.958 41.807 1.00 40.69 N
1959 ATOM 1961 C5 A A 62 22.523 15.840 42.134 1.00 45.39 C
1960 ATOM 1962 C6 A A 62 22.012 14.634 41.321 1.00 49.52 C
1961 ATOM 1963 N6 A A 62 22.234 14.614 40.078 1.00 44.46 N
1962 ATOM 1964 N1 A A 62 21.313 13.559 41.983 1.00 45.82 N
1963 ATOM 1966 C2 A A 62 21.087 13.586 43.394 1.00 45.05 C
1964 ATOM 1967 N3 A A 62 21.577 14.723 44.151 1.00 48.62 N
1965 ATOM 1968 C4 A A 62 22.304 15.843 43.481 1.00 49.84 C
1966 ATOM 1969 H5' A A 62 24.644 21.612 44.658 1.00 82.84 H
1967 ATOM 1970 H5'' A A 62 24.449 20.402 43.655 1.00 82.84 H
1968 ATOM 1971 H4' A A 62 22.998 20.672 46.060 1.00 70.64 H
1969 ATOM 1972 H3' A A 62 25.426 19.797 46.154 1.00 68.27 H
1970 ATOM 1973 H2' A A 62 25.087 18.142 44.574 1.00 61.99 H
1971 ATOM 1974 HO2' A A 62 25.201 17.142 46.957 1.00 62.91 H
1972 ATOM 1975 H1' A A 62 22.686 17.458 45.912 1.00 65.13 H
1973 ATOM 1976 H8 A A 62 23.815 18.352 42.502 1.00 63.43 H
1974 ATOM 1981 H2 A A 62 20.636 12.891 43.817 1.00 54.16 H Now we move onto our ligand (residue name GUN). The atom names reported in the pdb are different than atom names we have in our parameter file.
[user@cluster] head -n22 GUN.lib
!!index array str
"GUN"
!entry.GUN.unit.atoms table str name str type int typex int resx int flags int seq int elmnt dbl chg
"N1" "na" 0 1 131072 1 7 -0.334523
"C1" "cc" 0 1 131072 2 6 0.097034
"N2" "nd" 0 1 131072 3 7 -0.523097
"C2" "ca" 0 1 131072 4 6 0.144003
"C3" "ca" 0 1 131072 5 6 0.558035
"O1" "os" 0 1 131072 6 8 -0.329698
"N3" "nb" 0 1 131072 7 7 -0.745822
"C4" "ca" 0 1 131072 8 6 0.884237
"N4" "nh" 0 1 131072 9 7 -0.890772
"N5" "nb" 0 1 131072 10 7 -0.693379
"C5" "ca" 0 1 131072 11 6 0.325075
"H1" "hn" 0 1 131072 12 1 0.347262
"H2" "h5" 0 1 131072 13 1 0.176097
"H3" "hn" 0 1 131072 14 1 0.381855
"H4" "hn" 0 1 131072 15 1 0.381855
"C6" "c3" 0 1 131072 16 6 -0.005320
"H5" "h1" 0 1 131072 17 1 0.075719
"H6" "h1" 0 1 131072 18 1 0.075719
"H7" "h1" 0 1 131072 19 1 0.075719We need to rename the heavy atoms in the pdb file to their corresponding names in the parameter file. In order to simplify this process we will first remove the protons within the residue. LEaP can easily add these back later with the correct names.
2169 HETATM 2175 N1 GUN A 69 20.435 11.931 36.172 1.00 40.72 N
2170 HETATM 2176 C1 GUN A 69 21.059 12.713 37.080 1.00 45.85 C
2171 HETATM 2177 N2 GUN A 69 21.000 12.094 38.277 1.00 37.65 N
2172 HETATM 2178 C2 GUN A 69 20.352 10.944 38.115 1.00 42.37 C
2173 HETATM 2179 C3 GUN A 69 19.965 9.819 39.086 1.00 43.44 C
2174 HETATM 2180 O1 GUN A 69 20.255 9.900 40.233 1.00 39.68 O
2175 HETATM 2181 N3 GUN A 69 19.246 8.676 38.614 1.00 43.96 N
2176 HETATM 2182 C4 GUN A 69 18.873 8.588 37.236 1.00 43.78 C
2177 HETATM 2183 N4 GUN A 69 18.150 7.445 36.734 1.00 40.36 N
2178 HETATM 2184 N5 GUN A 69 19.248 9.652 36.333 1.00 41.19 N
2179 HETATM 2185 C5 GUN A 69 19.996 10.845 36.806 1.00 45.72 C Barium and sodium ions, and a few water molecules were resolved crystallographically. These are not catalytically relevant, and for the sake of simplicity we will assume they are also not structurally relevant and remove them.
2180 HETATM 2191 BA BA A 70 31.906 8.390 45.892 0.60 24.18 BA
2181 HETATM 2192 BA BA A 71 34.813 -6.355 31.778 0.58 30.39 BA
2182 HETATM 2193 BA BA A 72 31.033 0.308 49.682 0.40 61.24 BA
2183 HETATM 2194 BA BA A 73 33.004 17.329 49.683 0.45 54.07 BA
2184 HETATM 2195 BA BA A 74 27.824 -1.912 53.472 0.40 46.29 BA
2185 HETATM 2196 BA BA A 75 32.282 24.572 53.839 0.48 69.02 BA
2186 HETATM 2197 BA BA A 76 0.279 22.083 36.141 0.75 84.35 BA
2187 HETATM 2198 BA BA A 77 18.560 14.938 32.414 0.56 62.27 BA
2188 HETATM 2199 BA BA A 78 8.384 17.597 38.313 0.76120.11 BA
2189 HETATM 2200 NA NA A 79 36.287 16.215 43.272 1.00 34.26 NA
2190 HETATM 2201 NA NA A 80 31.896 7.690 34.088 1.00 50.91 NA
2191 HETATM 2202 O HOH A 81 43.549 22.559 52.718 1.00 67.88 O
2192 HETATM 2203 O HOH A 82 46.224 26.856 56.524 1.00 69.56 O
2193 HETATM 2204 O HOH A 83 -0.607 23.932 34.220 1.00 62.80 O
2194 HETATM 2205 O HOH A 84 13.884 27.718 35.721 1.00 64.85 O
2195 HETATM 2206 O HOH A 85 15.463 4.299 36.801 1.00 47.72 O
2196 HETATM 2207 O HOH A 86 8.577 12.465 25.488 1.00 70.15 O
2197 HETATM 2208 O HOH A 87 27.735 18.894 46.490 1.00 40.64 O
2198 HETATM 2209 O HOH A 88 33.130 -4.678 33.799 1.00 32.49 O
2199 HETATM 2210 O HOH A 89 32.100 4.191 32.161 1.00 39.50 O
2200 HETATM 2211 O HOH A 90 26.286 0.200 32.199 1.00 50.23 O
2201 HETATM 2212 O HOH A 91 36.070 10.612 25.896 1.00 55.39 O
2202 HETATM 2213 O HOH A 92 25.235 17.366 32.641 1.00 57.43 O
2203 HETATM 2214 O HOH A 93 34.079 -14.419 28.448 1.00 52.99 O
2204 HETATM 2215 O HOH A 94 35.911 -4.317 32.231 1.00 33.16 O
2205 HETATM 2216 O HOH A 95 28.622 -6.340 30.512 1.00 55.39 O
2206 HETATM 2217 O HOH A 96 21.541 18.363 31.228 1.00 45.76 O
2207 HETATM 2218 O HOH A 97 39.960 0.108 40.497 1.00 46.79 O
2208 HETATM 2219 O HOH A 98 5.321 31.207 42.190 1.00 59.35 O
2209 HETATM 2220 O HOH A 99 -0.444 19.583 36.126 1.00 52.16 O
2210 HETATM 2221 O HOH A 100 28.698 0.541 51.368 1.00 59.76 O
2211 HETATM 2222 O HOH A 101 15.735 -0.430 36.539 1.00 55.39 O
2212 HETATM 2223 O HOH A 102 37.524 9.301 27.209 1.00 62.05 O
2213 HETATM 2224 O HOH A 103 28.598 21.370 47.356 1.00 48.57 O
2214 HETATM 2225 O HOH A 104 33.224 6.071 32.685 1.00 44.69 O
2215 HETATM 2226 O HOH A 105 28.951 21.487 44.742 1.00 56.37 O
2216 HETATM 2227 O HOH A 106 30.511 21.905 41.786 1.00 63.69 O
2217 HETATM 2228 O HOH A 107 33.481 24.017 45.493 1.00 47.89 O
2218 HETATM 2229 O HOH A 108 -3.444 23.781 35.501 1.00 60.89 O
2219 HETATM 2230 O HOH A 109 27.024 21.879 45.193 1.00 58.85 O
2220 HETATM 2231 O HOH A 110 32.792 -2.728 32.364 1.00 43.28 O
2221 HETATM 2232 O HOH A 111 30.418 23.355 48.435 1.00 55.87 O
2222 HETATM 2233 O HOH A 112 31.672 27.219 56.474 1.00 56.58 O# MTR1 clean pdb representations
display depthcue off
mol showrep top 0 on
# Ions and water (vmd thinks the ligand is solvent, so exclude it)
mol selection {solvent and (not resname GUN)}
mol representation CPK
mol addrep top
# Active site residues
mol selection {resid 9 44 62 69}
mol representation licorice
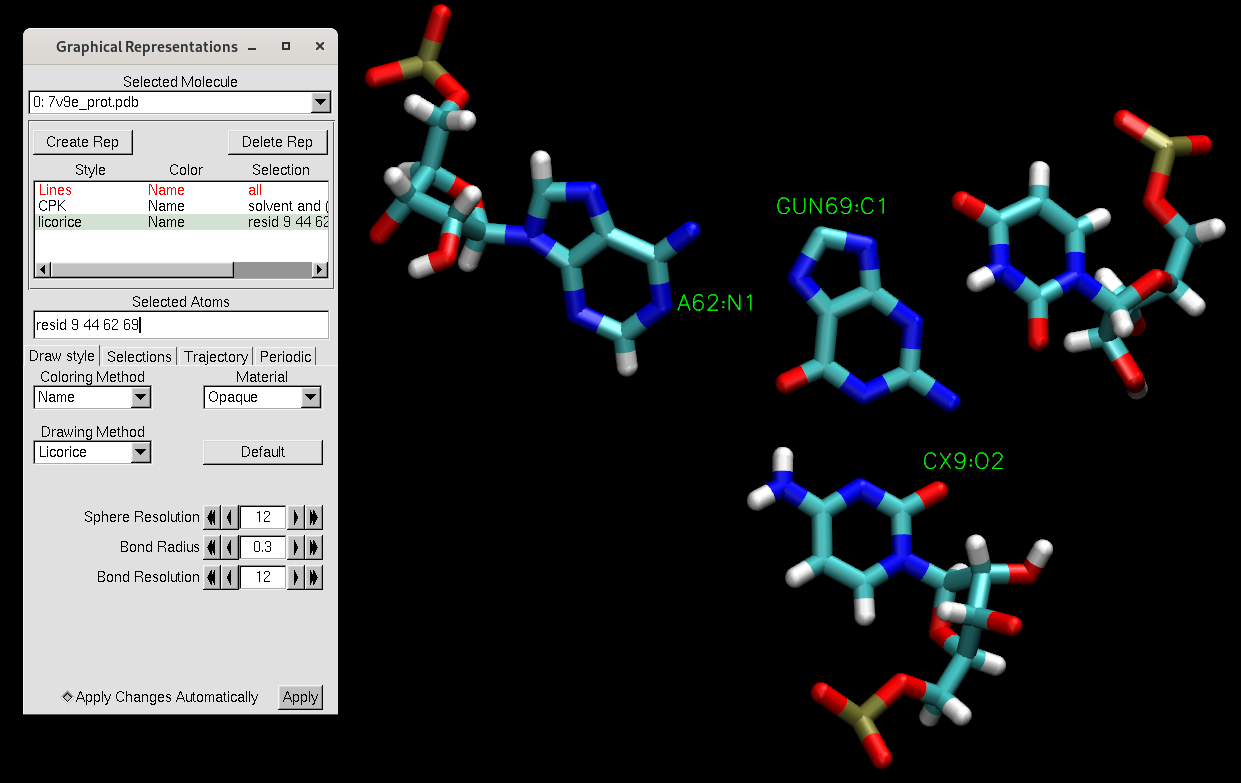
mol addrep top
There should be no solvent remaining. Since the pdb file has been reordered, the active site residues have different numbers from the file we downloaded from the PDB website. The active site representation now contains residues 9, 44, 62, and 69, which used to be residues 10, 45, 63, and 101.
AMBER has a program named LEaP, which takes in coordinates and force field definitions and produces parameter and restart files needed for system definition by the MD engine. We will use LEaP to create a simulation box that has explicit water molecules, Na+ and Cl- ions, as well as our RNA-ligand system in its protonated state. This process is also referred to as "solvating the system". LEaP can fill in missing atoms, but it will not delete atoms. This means the structure cannot contain atom types not included in a force field.
LEaP has a specific order of events. Force field parameters need to be loaded first, followed by loading the molecule. Next solvent and ions are added and finally output files need to be saved. Nucleic acids benefit from adding ions and water in a specific order. For pedagogical reasons, we will start with a step-wise approach to building the simulation box. At the end we will have a single input file that will build the simulation box in one go.
1. Force fields and loading the molecule:
In order for your molecule to be loaded without error, every residue and atom in your molecule file needs to have a corresponding parameter set already loaded to LEaP. AMBER has variety of established force fields for proteins and nucleic acids, as well as water models and metal ion parameters. Current recommendation for RNA is using OL3 (Zgarbová, 2011). We use TIP4P-Ew water (Horn, 2004) for nucleic acids systems, due to the availability of fine-tuned metal ion parameters which are often critical for nucleic acid structure and function.
Additionally our system has 2 non-standard residues (i.e. name not readily available within amber force fields), for which we need to use external parameters. In this tutorial they are already provided, you should have the parameter files CX.lib, CX.frcmod, GUN.lib, and GUN.frcmod. In upcoming tutorials you will learn how to parametrize these non-standard residues. First let's check if we have all the definitions we need. We will check by building restart and parameter files for the RNA-ligand alone without any solvent.
set default nocenter on # Do not recenter the molecule
### Sourcing amber forcefields ###############################################
source leaprc.RNA.OL3 # Sourcing RNA force field OL3 to describe RNA.
source leaprc.water.tip4pew # Sourcing water model TIP4P-Ew
# and parameters for corresponding monovalent ions
#loadamberparams frcmod.ions234lm_1264_tip4pew # divalent metal ion parameters
### Including non-standard residue parameters ################################
# Ligand O6 methylated guanine
loadoff GUN.lib
loadamberparams GUN.frcmod
# N3 protonated cytosine
loadoff CX.lib
loadamberparams CX.frcmod
### Loading structure from file ##############################################
mol= loadpdb "7v9e_prot.pdb"
### Saving output files #####################################################
# save parameter and restart files
saveamberparm mol MTR1_noSolvent.parm7 MTR1_noSolvent.rst7
quitRun tleap with tleap_MTR1_param.in as input:
[user@cluster] tleap -sf tleap_MTR1_param.inVisualize the resulting parameter and restart file in vmd. You may continue to use the MTR_cleanreps.txt until otherwise specified.
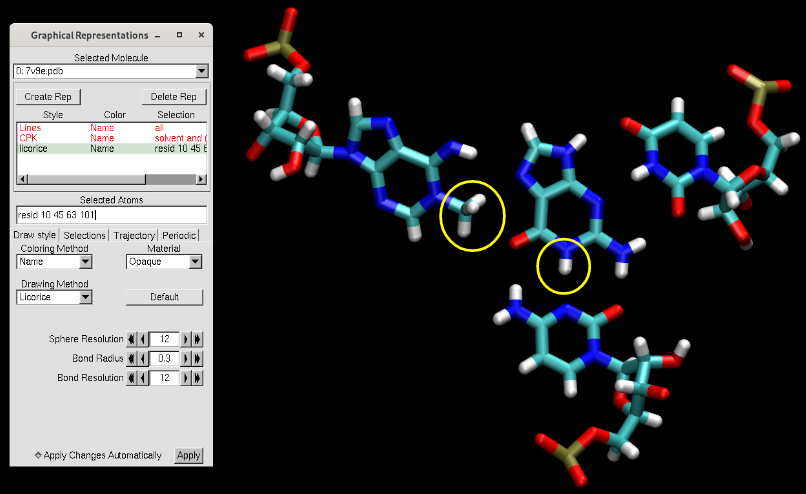
[user@local] vmd MTR1_noSolvent.parm7 MTR1_noSolvent.rst7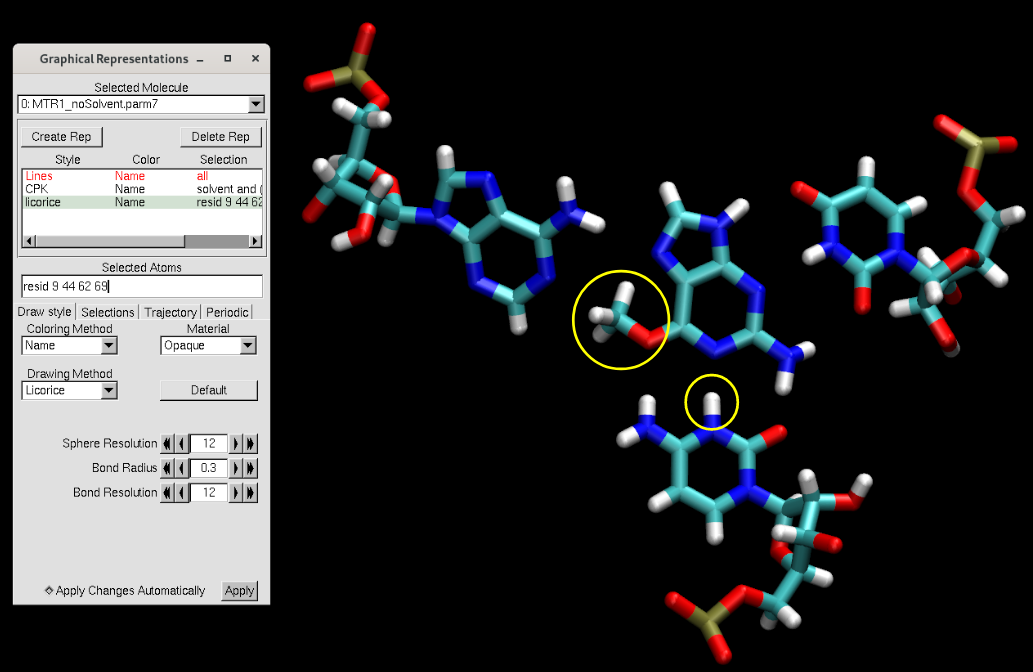
You will see that the methyl group was added on the ligand, and a proton was added onto the cytosine (circled in yellow above). Additionally, missing hydrogen atoms were added.
2. Neutralize the system with counterions
Now we will only add the counter ions to our system. Use the tleap_MTR1_counterIon.in input file, which differs from the above by only the inclusion of the following line and output file names.
### Adding counter ions ######################################################
addions mol NA 0 # Add sodium until charge is zero[user@cluster] tleap -f tleap_MTR1_counterIon.in[user@local] vmd MTR1_counterIon.parm7 MTR1_counterIon.rst7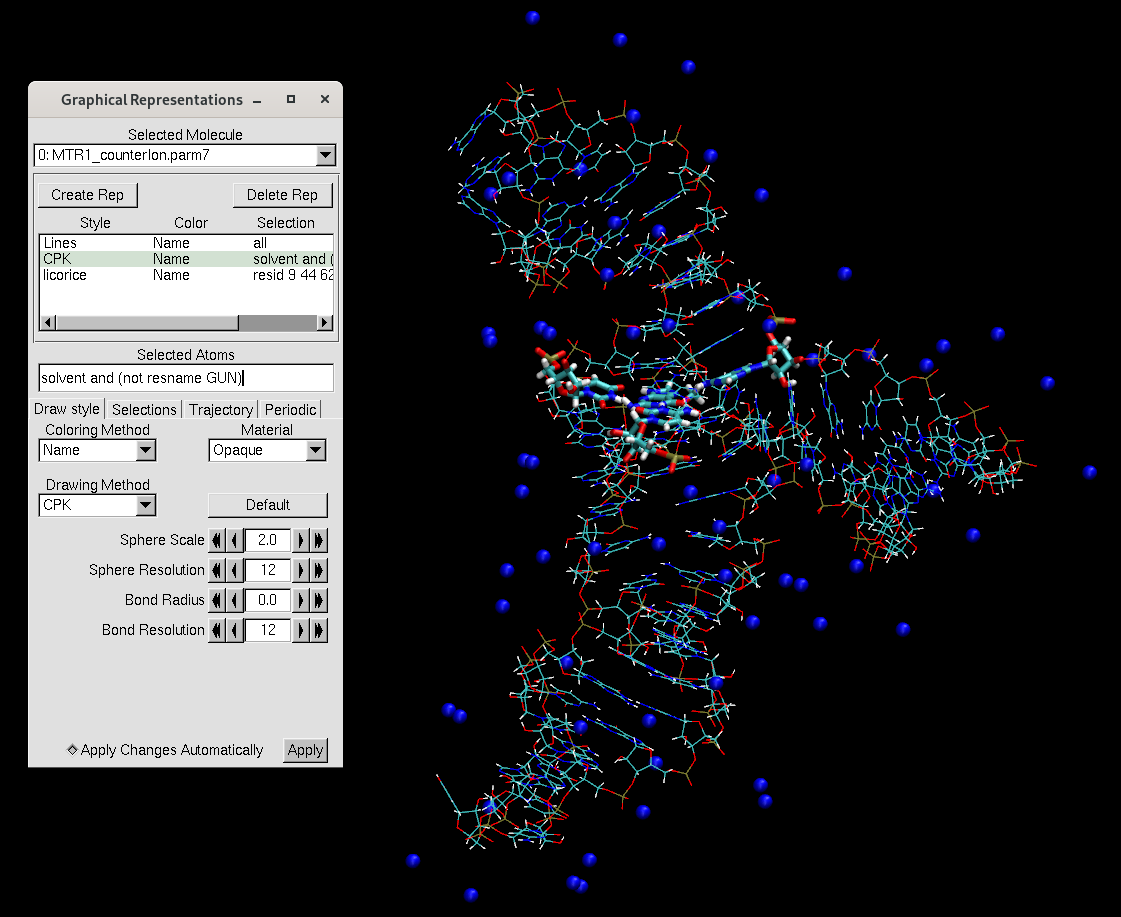
You will notice that the counterions are placed along the phosphate backbone, which is negatively charged. It is essential to neutralize the backbone to stabilize the structure. We add the counterions before adding water to avoid the ions being placed far away from the RNA.
3. Add water molecules
Next we will add water molecules. The simulations will ultimately take place under periodic boundary conditions, but we will must define the shape of the box for a given image. Since the RNA is somewhat globular, we will chose a truncated octahedron. This is closer to a sphere than say, a rectangular box. The size of the box will define the volume of the system. We want to create a sufficient buffer of solvent around the RNA, so we will use a solvation radius of 9 Å. We will use a 4-point water model.
Now we will add the water to our system. Use the tleap_MTR1_counterIon_water.in input file, which differs from the above by only the inclusion of the following line and output file names.
### Defining box shape & size with chosen water model ########################
solvateoct mol TIP4PEWBOX 9 # solvate with truncated octahedron box 9A out[user@cluster] tleap -f tleap_MTR1_counterIon_water.in[user@local] vmd MTR1_counterIon_water.parm7 MTR1_counterIon_water.rst7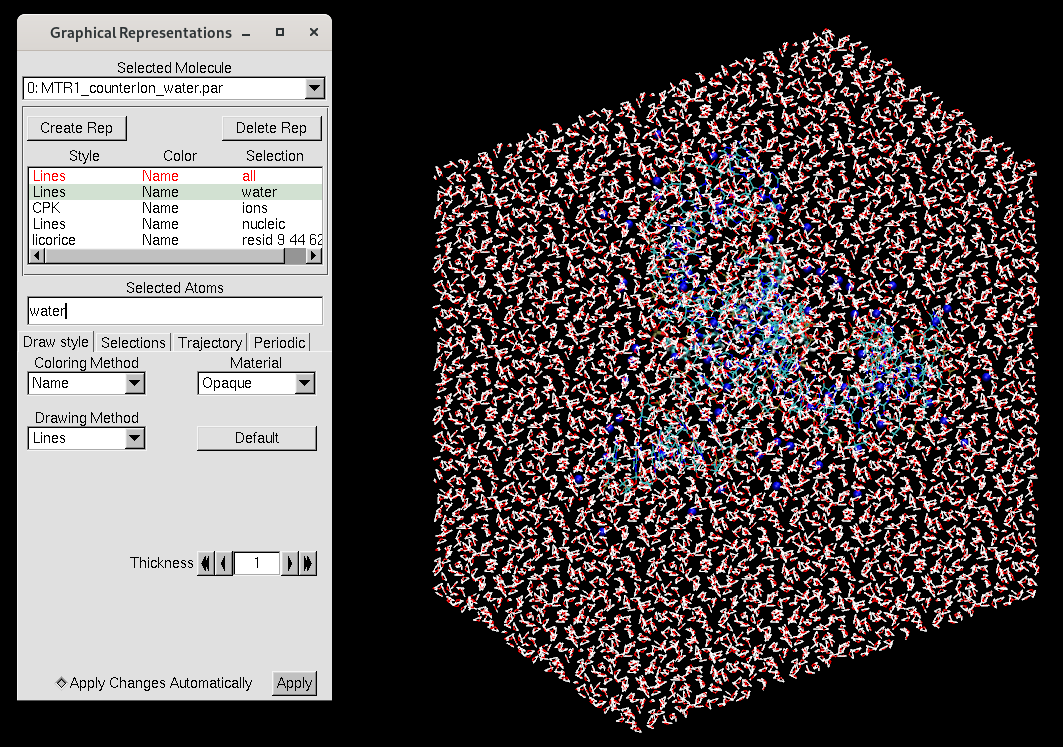
The representations now seperate the water and ions rather than referring to just solvent. Here are the contents of MTR_waterreps.txt:
# MTR1 solvated representations
display depthcue off
mol showrep top 0 off
# water
mol selection {water}
mol representation Lines
mol addrep top
# Ions (vmd thinks the ligand is solvent, so exclude it)
mol selection {ions}
mol representation CPK 2.0
mol addrep top
# RNA
mol selection {nucleic}
mol representation Lines
mol addrep top
# Active site residues
mol selection {resid 9 44 62 69}
mol representation licorice
mol addrep top4. Add bulk ions
Next you will add bulk Na+ and Cl- ions to the water box to achieve a bulk concentration of 0.14 M. This is meant to mimic physiological ion concentration. Based on the box volume established in the last step, we will need to add an additional 45 NaCl. These will be placed at least 6 Å away from each other. The following has been added to the input file:
## Adding bulk ions to ensure 0.14 M concentration ##########################
addionsrand mol NA 45 CL 45 6.0 # add bulk ions, replacing random waters,
# ensuring 6 A distance between ionsInclusion of this line completes everything we wanted to do to build our box. Contents of our stand-alone input file are:
set default nocenter on # Do not recenter the molecule
### Sourcing amber forcefields ###############################################
source leaprc.RNA.OL3 # Sourcing RNA force field OL3 to describe RNA.
source leaprc.water.tip4pew # Sourcing water model TIP4P-Ew
# and parameters for corresponding monovalent ions
#loadamberparams frcmod.ions234lm_1264_tip4pew # divalent metal ion parameters
### Including non-standard residue parameters ################################
# Ligand O6 methylated guanine
loadoff GUN.lib
loadamberparams GUN.frcmod
# N3 protonated cytosine
loadoff CX.lib
loadamberparams CX.frcmod
### Loading structure from file ##############################################
mol= loadpdb "7v9e_prot.pdb"
### Adding counter ions ######################################################
addions mol NA 0 # Add sodium until charge is zero
### Defining box shape & size with chosen water model ########################
solvateoct mol TIP4PEWBOX 9 # solvate with truncated octahedron box 9A out
### Adding bulk ions to ensure 0.14 M concentration ##########################
addionsrand mol NA 45 CL 45 6.0 # add bulk ions, replacing random waters,
# ensuring 6 A distance between ions
### Saving output files #####################################################
# save parameter and restart files
saveamberparm mol MTR1_counterIon_water_bulk.parm7 MTR1_counterIon_water_bulk.rst7
savepdb mol MTR1_counterIon_water_bulk_amber.pdb
quit
We will use this final input file to build our simulation box.
[user@cluster] tleap -sf MTR1_counterIon_water_bulk.inThis will generate MTR1_counterIon_water_bulk.parm7 and MTR1_counterIon_water_bulk.rst7 that will be used to perform MD simulations.
[user@local] vmd MTR1_counterIon_water_bulk.parm7 MTR1_counterIon_water_bulk.rst7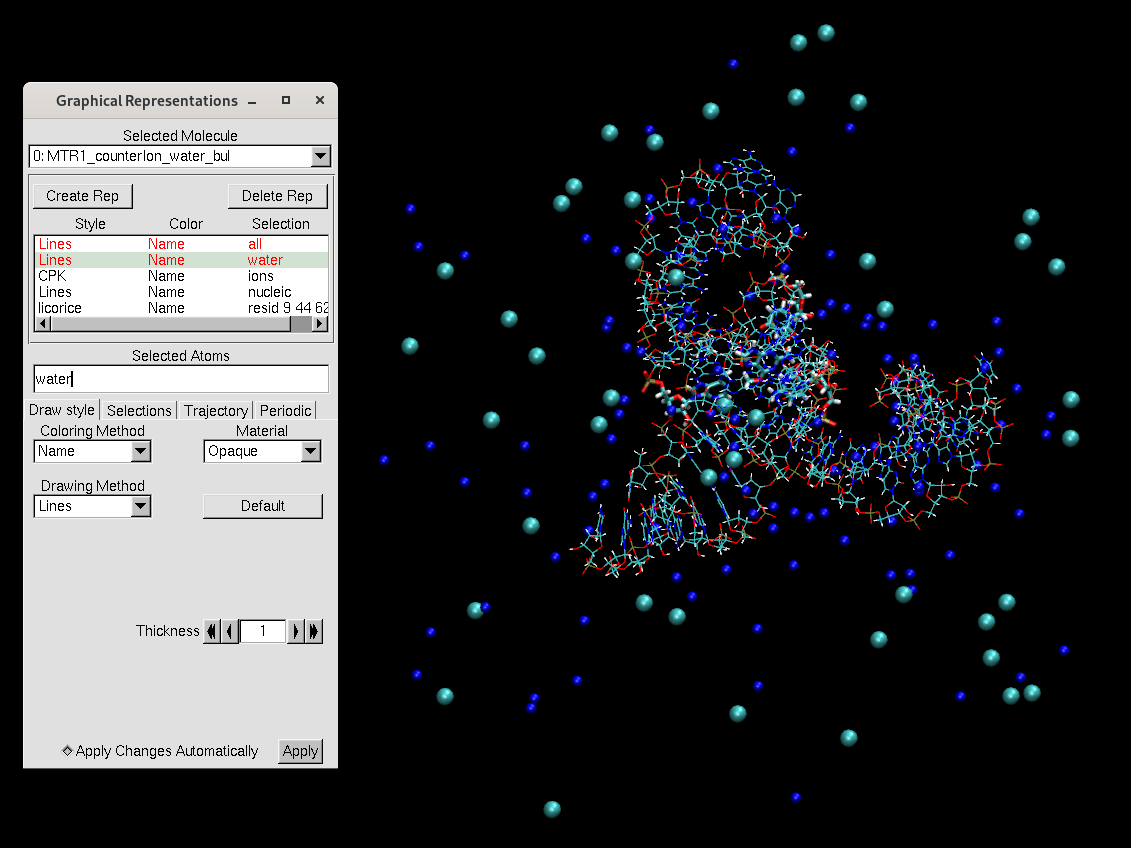
The necessary inputs and subsequent outputs for parts 1.3, 1.4, and 1.5 are available in:
Files/input/Equil
and
Files/output/Equil
The inputs should be copied to your working directory. If you unable to produce an output for a given step, copy it from the output directory in order to proceed.
[user@cluster] mkdir MTR1_Equil
[user@cluster] cd MTR1_Equil
[user@cluster] cp -r Path2/Files/input/Equil/* ./ Now you will learn how to equilibrate the simulation box. Electrostatic interactions are extremely important for RNA structure given the negatively charged backbone. Therefore, a rigorous equilibration process allows the solvent to properly organize around the molecule in a controlled way while avoiding large fluctuations in density that could cause errors. This is especially important when your system contains divalent metal ions! The electrostatic interaction energy is proportional to the product of the two charges, so increasing the charge will lead to a larger interaction energy at a given distance. Nucleic acids are highly charged systems and must be equilibrated slowly. You have been provided the Equil directory containing eqFromStratch.slurm and a directory called mdins.
[user@cluster] ls mdins
000_Min.mdin 003_NPT.mdin 03_Equil_NPT_solvent.mdin 06_Equil_solute_Rwt25.mdin 09_Equil_solute_Rwt2.mdin
001_NPT.mdin 01_Min.mdin 04_Min_solute.mdin 07_Equil_solute_Rwt10.mdin 11_Production_0.5ns.mdin
002_NPT.mdin 02_Heat.mdin 05_Heat_solute_Rwt25.mdin 08_Equil_solute_Rwt5.mdinThese are the input files for the stepwise equilibration procedure and subsequent production simulation. Here is a summary of the equilibration steps:
The general approach is to slowly equilibrate the solvent around the solute, then gradually allow the solute to move. We cycle through minimizations to remove clashes and find a local minimum, heating to explore new conformations, then simulations in the NPT ensemble to approach the global minimum. There are some specific points to note about how the provided mdin files have been written.
Initial Solvent Energy Minimization Stage
Bulk water and Sodium ions only
&cntrl
! Run Control
imin = 1, ! Flag to run minimization
ntmin = 0, ! Minimization algorithm
maxcyc = 500, ! maximum # of cycles for minimization
! Input_Output Control
ntx = 1, ! Read coordinates, but not velocities
ntpr = 250, ! Every ntpr steps, print human-readable energy info
ntwx = 0, ! Every ntwx steps, print coordinates to mdcrd file
! Restraints
ntr = 1, ! Flag for restraining specified atoms
restraint_wt = 50.0, ! Restraint weight (in kcal/mol-A2) for positional
restraintmask = '!@H= & !:NA,CL,MG,WAT', ! (restrain only solute)
! Miscellaneous
cut = 12.0, ! Non-bonded interaction cutoff (in A)
/
&wt type = 'DUMPFREQ', istep1 = 10000, /
&wt type='END' /
DISANG=restrnt.disang
DUMPAVE=restrnt.dumpave
LISTOUT=POUT
LISTIN=POUTThe yellow highlighted line indicates that restraints will be applied to all atoms not (!) included in the mask @H= and not :NA,CL,MG,WAT. This means all heavy atoms that are not sodium, chloride, magnesium, or water will experience the positional restraint (ie. the RNA heavy atoms are effectively frozen in place). There is no Mg2+ in this system currently, but many RNA systems will contain it. If your system includes other solvent atoms, they should be included in this mask. When we added ions in LEaP we referred to them as NA and CL, but one could also have said Na+ and Cl-, which would also be specified in the mask.
The green highlighted line means that a file called restrnt.disang will be read as input. This file could contain nmr restraints between specified atoms such as a distance, angle, or torsion. This is different than coordinate restraints invoked by setting ntr = 1, as the atom positions may fluctuate while adhering to the restraint relative to the other atoms. A common example use case could be maintaining an important hydrogen bond that could be lost during equilibration. You will learn more about using nmr type restraints in a future session. For the sake of this tutorial, no nmr restraints will be applied; however this line is maintained in our workflow because it allows us to have one centralized copy of the mdin files regardless of whether additional restraints are desired. In this case, restrnt.disang will be empty.
[user@cluster] touch restrnt.disang Now we will review how the simulations are run. You have been provided eqFromStratch.slurm.
#!/bin/bash
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.err
#SBATCH --nodes=1
#SBATCH --gpus=1
#SBATCH --ntasks-per-node=1
#SBATCH --partition=gpu-shared
#SBATCH --account=gue998
#SBATCH --reservation=amber24gpu
#SBATCH -t 2-00:00:00
#SBATCH --no-requeue
module load workshop/amber24/default
set -x
echo AMBERHOME is $AMBERHOME
test -f ${AMBERHOME}/amber.sh && source ${AMBERHOME}/amber.sh
LAUNCH="srun --kill-on-bad-exit"
EXE=${AMBERHOME}/bin/pmemd.cuda
BASE=$1
PARM=${BASE}.parm7
REF=${BASE}.rst7
mdins=mdins
NAME=${BASE}
steps="\
000_Min \
001_NPT \
002_NPT \
003_NPT \
01_Min \
02_Heat \
03_Equil_NPT_solvent \
04_Min_solute \
05_Heat_solute_Rwt25 \
06_Equil_solute_Rwt25 \
07_Equil_solute_Rwt10 \
08_Equil_solute_Rwt5 \
09_Equil_solute_Rwt2 \
"
#switch for skipping existing steps
k=1
for step in $steps; do
ICRD=${BASE}.rst7
MDIN=${mdins}/${step}.mdin
BASE=${step}-run
checkMDout1=$(tail -n 1 ${BASE}.mdout 2>/dev/null | grep "Total wall time")
echo "Check 1 is $checkMDout1"
checkMDout2=$(grep "wallclock() was called" ${BASE}.mdout)
echo "Check 2 is $checkMDout2"
if [ "$k" == 1 ]; then
if [ -f "${BASE}.mdout" ] && [ ! -z "$checkMDout1" ]; then
echo "${BASE} step complete, skipping"
continue;
elif [ -f "${BASE}.mdout" ] && [ ! -z "$checkMDout2" ]; then
echo "${BASE} step complete, skipping"
continue;
else
k=0
echo "Starting from step ${step} and not skipping"
fi
fi
if [ "$step" == 000_Min ] || [ "$step" == 01_Min ]; then
restartfile=${BASE}.rst7
trajFile=${BASE}.rst7
EXE=${AMBERHOME}/bin/sander
else
restartfile=${BASE}.rst7
trajFile=${BASE}.nc
EXE=${AMBERHOME}/bin/pmemd.cuda
fi
echo running ${MDIN}
${LAUNCH} ${EXE} -O -p ${PARM} -c ${ICRD} -i ${MDIN} -o ${BASE}.mdout -inf ${BASE}.mdinfo -x ${BASE}.nc -r ${BASE}.rst7 -ref ${REF}
mv ${BASE}.mdinfo ${BASE}.stat
echo "MDOUT IS ${BASE}.mdout"
checkMDout1=$(tail -n 1 ${BASE}.mdout 2>/dev/null | grep "Total wall time")
echo "Check 1 is $checkMDout1"
checkMDout2=$(grep "wallclock() was called" ${BASE}.mdout)
echo "Check 2 is $checkMDout2"
if [ -f "${BASE}.mdout" ] && [ ! -z "$checkMDout1" ]; then
echo "Check 1 is $checkMDout1"
echo "${BASE} step complete, moving on"
continue;
elif [ -f "${BASE}.mdout" ] && [ ! -z "$checkMDout2" ]; then
echo "Check 2 is $checkMDout2"
echo "${BASE} step complete, moving on"
continue;
else
echo "${BASE} did not finish correctly, exiting."
exit 1
fi
doneThis script will takes as input the prefix of the parameter and restart files. The yellow highlighted lines refer to the equilibration steps. They will be run one at a time. If a step fails, one can easily pick up at the failed step and skip the steps that have already finished. The green highlighted lines indicate that the early minimization steps will be performed with sander, and the rest of the steps will be performed with pmemd.cuda. This is because pmemd.cuda is more sensitive to large spikes in the energy than sander, and we want to avoid errors.
The duration of the equilibration procedure is too long for this workshop; however, the job would be submitted as follows:
[user@cluster] sbatch eqFromStratch.slurm MTR1_counterIon_water_bulkThe outputs of the equilibration procedure have been provided for you in the Files/output/Equil directory. Now we will compare the structure from before and after equilibration. The last step of equilibration produced output with the prefix 09_Equil_solute_Rwt2-run. Now that we have performed simulations under periodic boundary conditions, atoms may have diffused into neighboring images. To visualize a single periodic unit, we need to wrap the solvent around the RNA, which is a concept you have been introduced to in a previous session. You have been provided a cpptraj input file called wrap_RNA_equil.in.
[user@cluster] cp Path2/Files/output/Equil/0*-run* ./ parm MTR1_counterIon_water_bulk.parm7
trajin 09_Equil_solute_Rwt2-run.rst7
reference 09_Equil_solute_Rwt2-run.rst7
center '!@H= & !:MG= & !:NA= & !:CL= & !:WAT=' mass origin
autoimage '!@H= & !:MG= & !:NA= & !:CL= & !:WAT='
center '!@H= & !:MG= & !:NA= & !:CL= & !:WAT=' mass reference
trajout centered.09_Equil_solute_Rwt2-run.rst7
run
quitIn this input file we set the center of mass of the RNA at the origin, then we image the solute around the RNA. Finally we center the imaged system based on the reference restart file and output a new restart file with the prefix "centered" to distinguish it. Now we can inspect the wrapped structure in vmd.
[user@cluster] cpptraj -i wrap_RNA_equil.invmd -f MTR1_counterIon_water_bulk.parm7 centered.09_Equil_solute_Rwt2-run.rst7 -f MTR1_counterIon_water_bulk.parm7 MTR1_counterIon_water_bulk.rst7 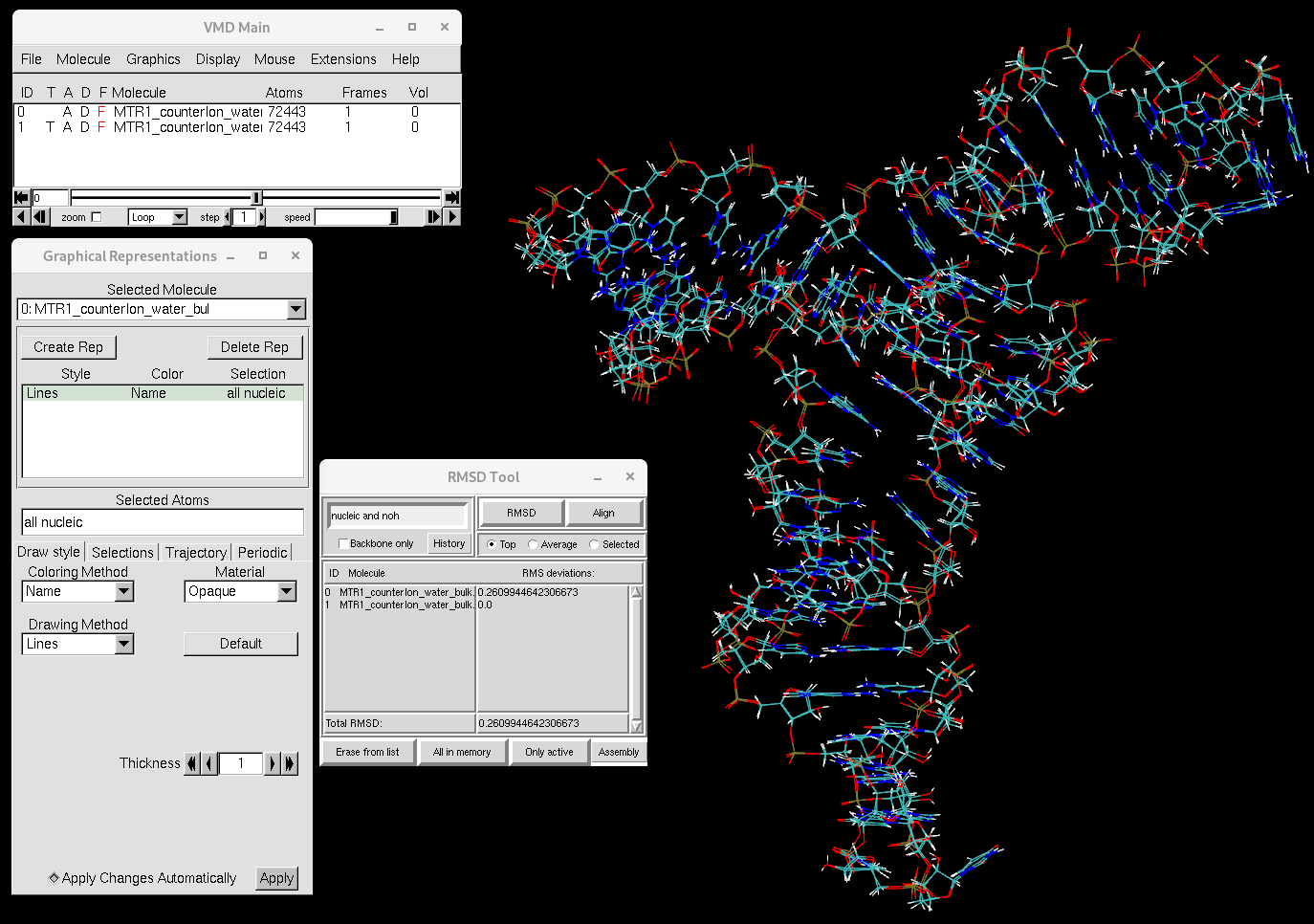
You will see that the RNA structure has not changed much over the course of equilibration. Now let's focus on the ions.
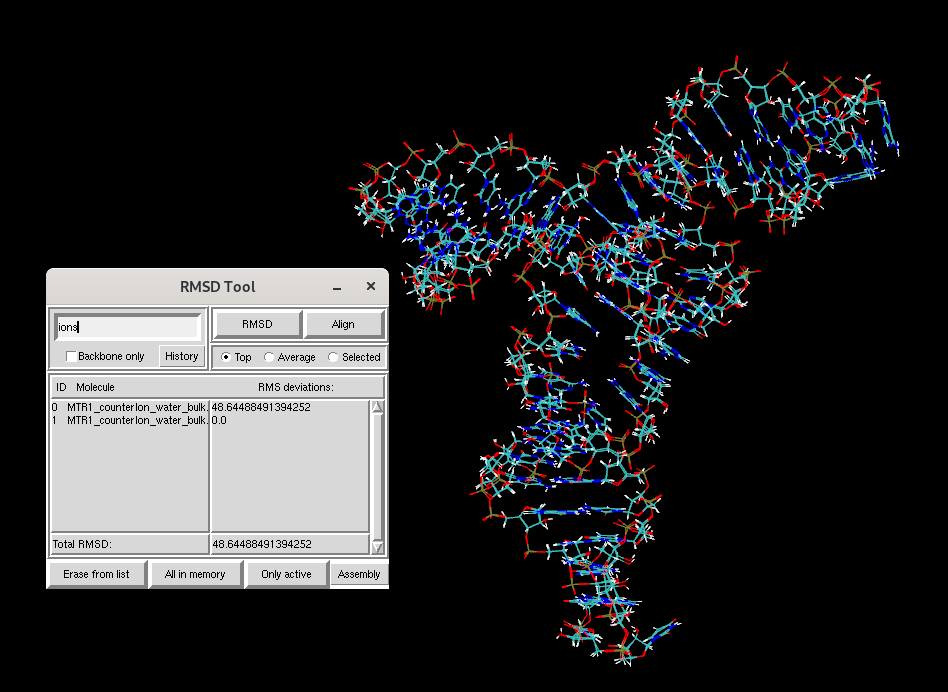
You will see that the RMSD for the ions between the two structures is much larger than that of the RNA. To study the dynamics of the RNA, you will next learn to run production MD simulations starting from your equilibrated structure
Next you will run a very short 0.5 ns production simulation where there are no restraints on the RNA. In previous studies of this RNA system, production simulations converged on the order of 50 ns, but although longer time scales are desirable, such simulations cannot be performed under the time constraints of the workshop. In the mdins directory you have been provided 11_Production_0.5ns.mdin. This will perform 0.5 ns of unrestrained dynamics in the isothermal-isobaric ensemble (NPT) to give you a quick introduction to running production simulations. You have also been provided the slurm script prod.slurm.
#!/bin/bash
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.err
#SBATCH --nodes=1
#SBATCH --gpus=1
#SBATCH --ntasks-per-node=1
#SBATCH --partition=gpu-shared
#SBATCH --account=gue998
#SBATCH --reservation=amber24gpu
#SBATCH -t 0-00:30:00
#SBATCH --no-requeue
Base=${1}
Parm=${1}.parm7
mdin=mdins/11_Production_0.5ns.mdin
module load workshop/amber24/default
newname=""
if [[ $Base == *-run-* ]]; then
prefix=${Base%-*} # Extract the part before the last hyphen
suffix=${Base##*-} # Extract the part after the last hyphen
if [[ $suffix =~ [0-9]+ ]]; then
number=${suffix#*-} # Extract the numeric part after the second hyphen
new_number=$(printf "%02d" $((number + 1))) # Increment the number and pad with leading zeros if needed
newname="${prefix}-${new_number}"
else
newname="${Base}-01"
fi
else
newname="${Base}-01"
fi
path=`pwd`
exe=${AMBERHOME}/bin/pmemd.cuda_SPFP
$exe -O -p ${Parm} -c ${Base}.rst7 -i ${mdin} -o ${newname}.mdout -inf ${newname}.mdinfo -x ${newname}.nc -r ${newname}.rst7 -ref ${Base}.rst7This script will take as an argument the prefix of the parm and restart file, which should be the same. It then takes that prefix and adds -01 to it. Although you are only running a short production simulation, it is often the case that longer simulations must be broken up into multiple runs due to time limits on an HPC. If the input prefix already has a number because you are restarting a simulation, it will increment to -02 and so on.
[user@cluster] cp 09_Equil_solute_Rwt2-run.rst7 MTR1_counterIon_water_bulk-run.rst7We will run production with a 4 fs timestep. In order to do this stably, we will use hydrogen mass repartitioning. You have been provided hmr.in that will repartition MTR1_counterIon_water_bulk.parm7 and output MTR1_counterIon_water_bulk-run.parm7.
cpptraj -i hmr.in[user@cluster] sbatch prod.slurm MTR1_counterIon_water_bulk-run The job should take about 10 minutes to complete. If you are waiting, consider attempting the additional exercise. The job will generate output with the prefix MTR1_counterIon_water_bulk-run-01. Once the job is done, we will look at the generated trajectory (.nc). You have been provided the cpptraj input file wrap_RNA_prod.in. This only differs from wrap_RNA_equil.in in that it now reads in the restart file MTR1_counterIon_water_bulk-run-01.rst7 as a reference and wraps MTR1_counterIon_water_bulk-run-01.nc.
[user@cluster] cpptraj -i wrap_RNA_prod.inThis will produce centered.MTR1_counterIon_water_bulk-run-01.nc. This structure can now be visualized in vmd.
[user@local] vmd centered.MTR1_counterIon_water_bulk-run-01.nc MTR1_counterIon_water_bulk-run.parm7
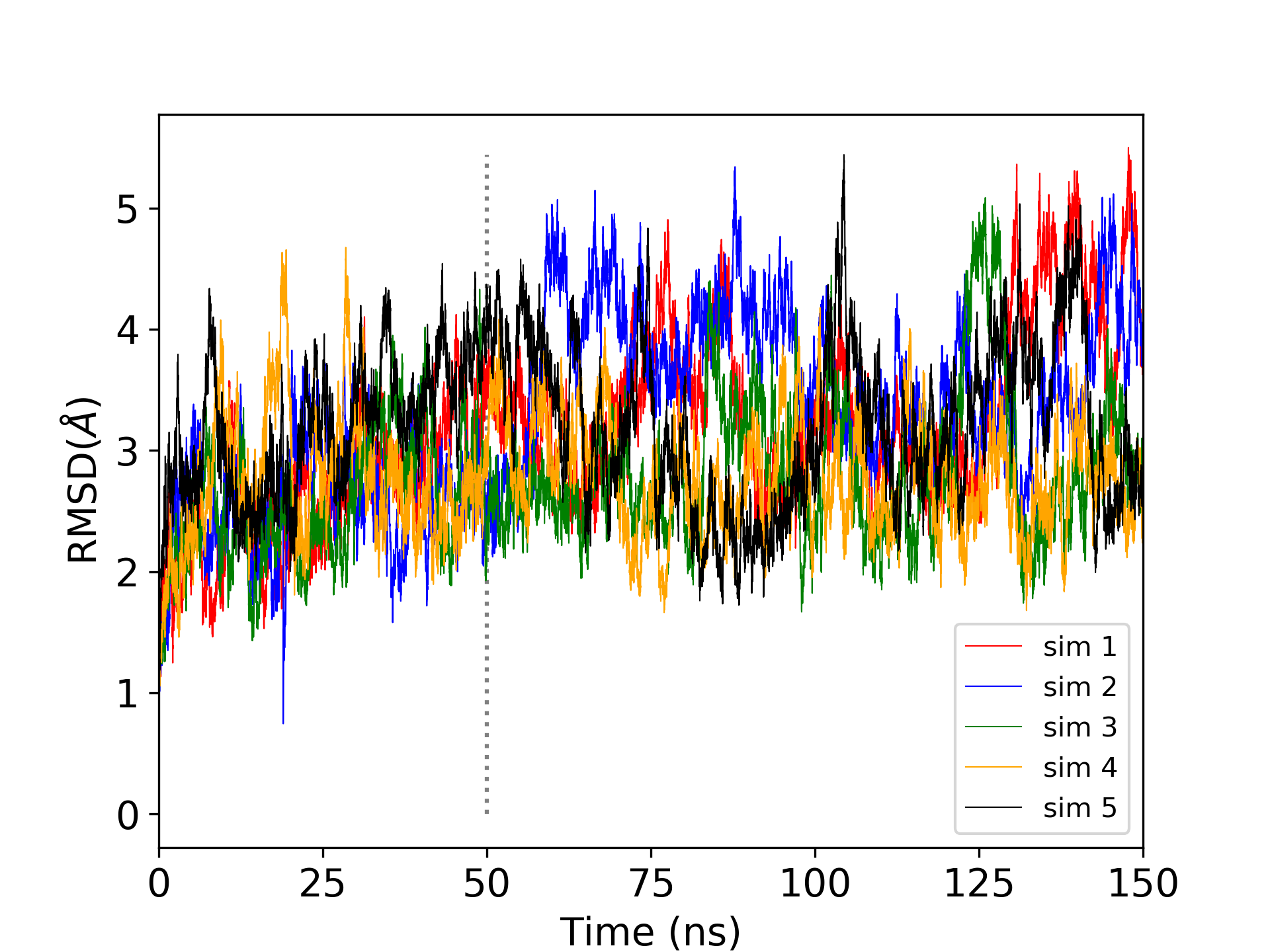
This was a very short production simulation. Realistically, it would be desirable to perform multiple trials of long production simulations. 5 trials of 150 ns production simulations were performed for MTR1, and the root mean square deviation was analyzed. The grey vertical line indicates that the first 50 ns were considered equilibration as the RMSD is increasing, so subsequent analysis was performed on the last 100 ns (also called the production region) of each simulation totaling 500 ns.
The following exercise should only be performed if you have time left over or are waiting for other simulations to run. In this exercise you will run the 000_Min step of equilibration.
[user@cluster] cp eqFromStratch.slurm eqFromStratch_000.slurm #!/bin/bash
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.err
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=1
#SBATCH --partition=shared
#SBATCH --account=gue998
#SBATCH --reservation=amber24
#SBATCH -t 0-00:30:00
#SBATCH --no-requeuesteps="\
000_Min \
"In this case we will only be invoking sander, so we do not need to request a gpu. The job is expected to finish in ~10 minutes.
The Equil directory should contain the following (if you are also running production simulations in the same directory from the previous exercise, that is ok. If you copied 000_Min.mdout from output already, you may need to delete it, otherwise the script will see that it already ran and quit):
[user@cluster] ls
MTR1_counterIon_water_bulk.parm7 MTR1_counterIon_water_bulk.rst7 eqFromStratch.slurm eqFromStratch_000.slurm mdins restrnt.disang[user@cluster] sbatch eqFromStratch_000.slurm MTR1_counterIon_water_bulk [user@cluster] grep -r EAMBER 000_Min-run.mdout | awk '{print $3}' | head -n10 > ene.dat[user@cluster] xmgrace ene.dat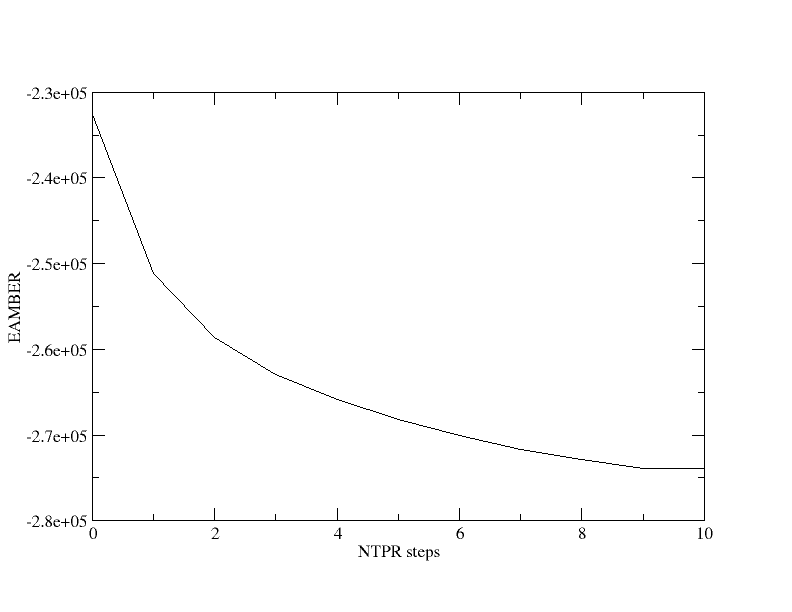
You will see that the total energy in kcal/mol decreases rapidly and begins to plateau. In this run we printed the energy energy 50 steps.
NSTEP ENERGY RMS GMAX NAME NUMBER
1 1.7197E+06 6.7827E+04 2.2501E+07 N3 1730
BOND = 5038.2053 ANGLE = 397.2193 DIHED = 1511.0059
VDWAALS = 1926187.1298 EEL = -206390.2786 HBOND = 0.0000
1-4 VDW = 697.4115 1-4 EEL = -7778.5268 RESTRAINT = 0.0000
NSTEP ENERGY RMS GMAX NAME NUMBER
50 -2.3201E+05 4.6787E+00 6.6797E+02 C6 1723
BOND = 22954.6607 ANGLE = 746.4316 DIHED = 1649.0907
VDWAALS = 27936.2925 EEL = -278521.9124 HBOND = 0.0000
1-4 VDW = 650.9327 1-4 EEL = -7954.8135 RESTRAINT = 533.4714
EAMBER = -232539.3177You will notice that the Van der Waals energy decreases rapidly in the beginning as bad contacts are alleviated. This should give you a sense of the importance of a gradual equilibration procedure.